DISCNGINE BLOG
Insights on drug discovery trends, events, product updates, use cases, tech workflows, customer stories, and life at Discngine.
Categories

Patent exploration with Ideation Analytics: The Paltusotine use case
On the Crinetics Therapeutics’ recently approved drug example we performed retroactive study with Ideation Analytics, showing how it supports patent exploration through efficient functionality and high-quality visualizations that follow medicinal chemists' workflows.

What are the common directions shaping modern antibody discovery? – Reflections from antibody events
Our Biologics Solution Lead, Méliné Simsir, has compiled insights from four recent antibody conferences she has attended, bringing together perspectives from experts across the community.
Designing with novelty: why considering Structure-Activity Relationship alongside patent disclosures matters
During lead optimization, SAR analysis drives compound design, while patent data provides context on prior art. These perspectives are often reviewed separately, making early decisions harder. This article looks at how bringing them together in a single workflow with tools such as Discngine Ideation Analytics, reduces rework and guides more confident design choices.
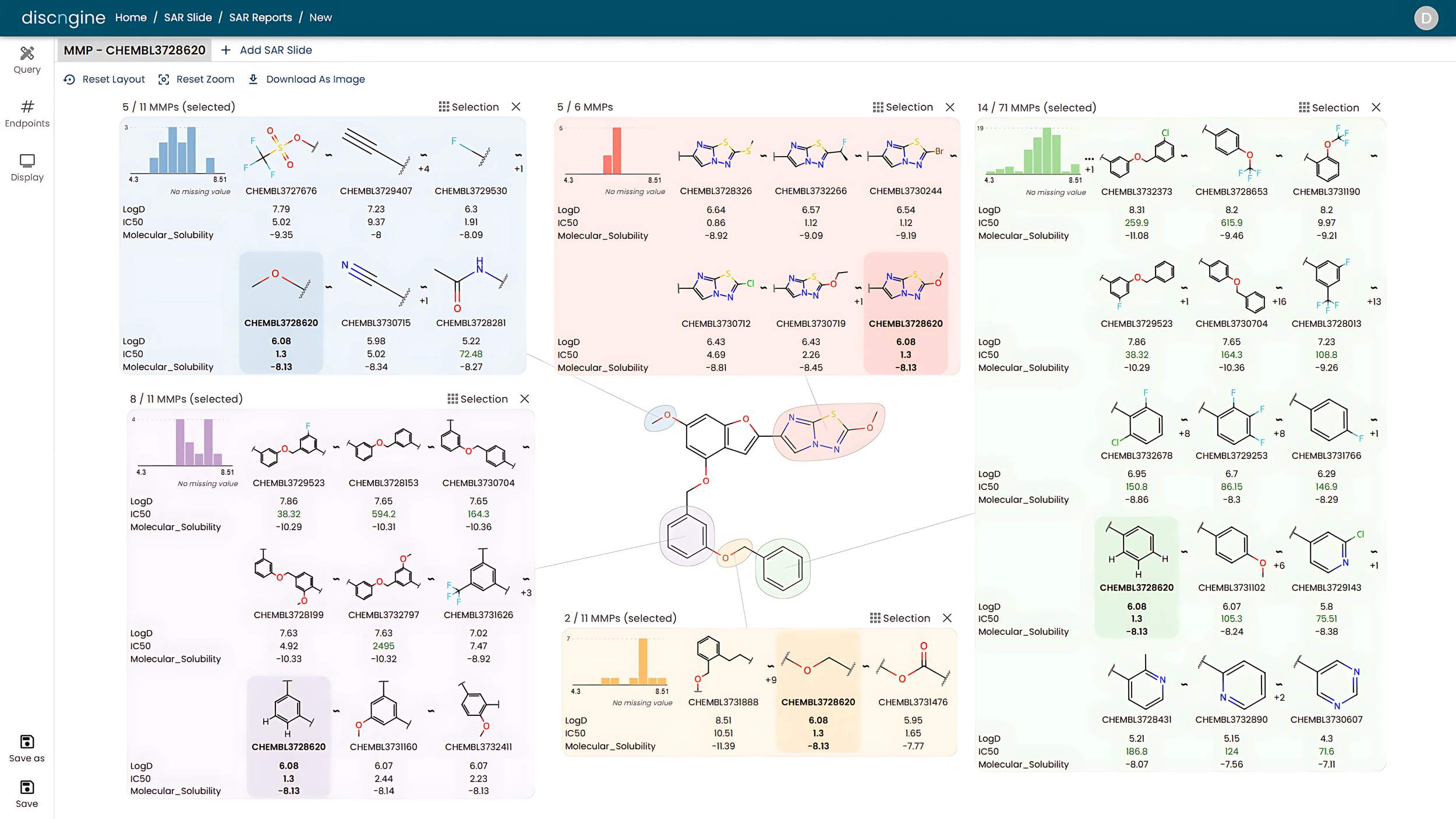
Efficient Structure-Activity Relationship (SAR) Reporting and Exploration in Drug Discovery: Discover Discngine Ideation Analytics
Manual structure-activity relationship (SAR) reporting is a time-consuming, tedious, and repetitive task.
To regain time from data wrangling, Discngine has developed Ideation Analytics- an automated platform for SAR reporting and analysis.

Ideation Analytics Gets Patent Searches with GOSTAR™ Integration
To streamline the Structure-Activity Relationship (SAR) exploration of patents, it is now possible to access GOSTAR™ patent datasets directly within Ideation SAR Slides.
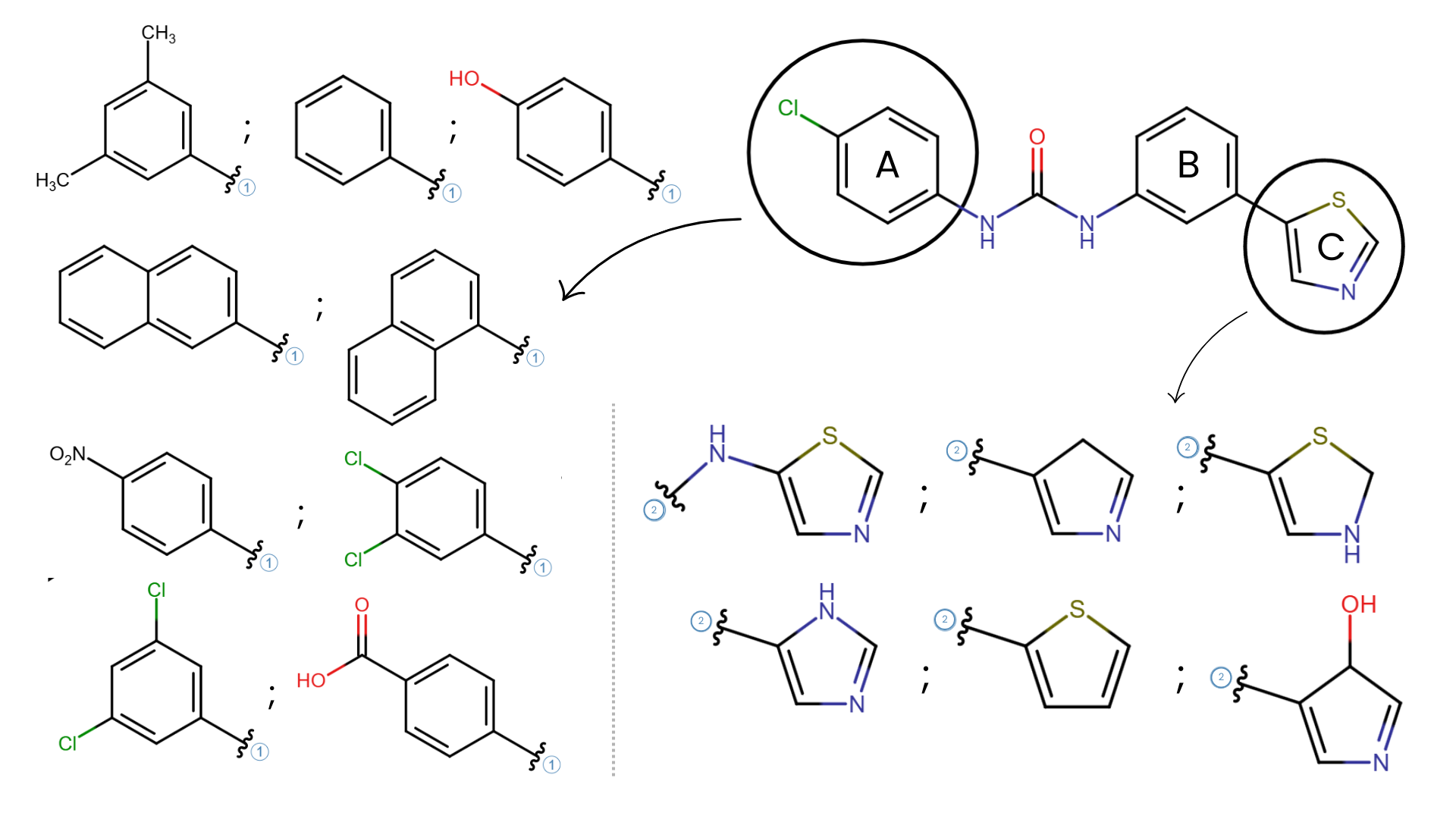
The 3 Hidden Costs of Manual SAR Reporting in Drug Discovery
Structure–activity relationship (SAR) reporting is central to small-molecule drug discovery, yet many teams still rely on manual workflows. This article explores the limitations of manual SAR reporting and how they affect DMTA cycles and decision-making.
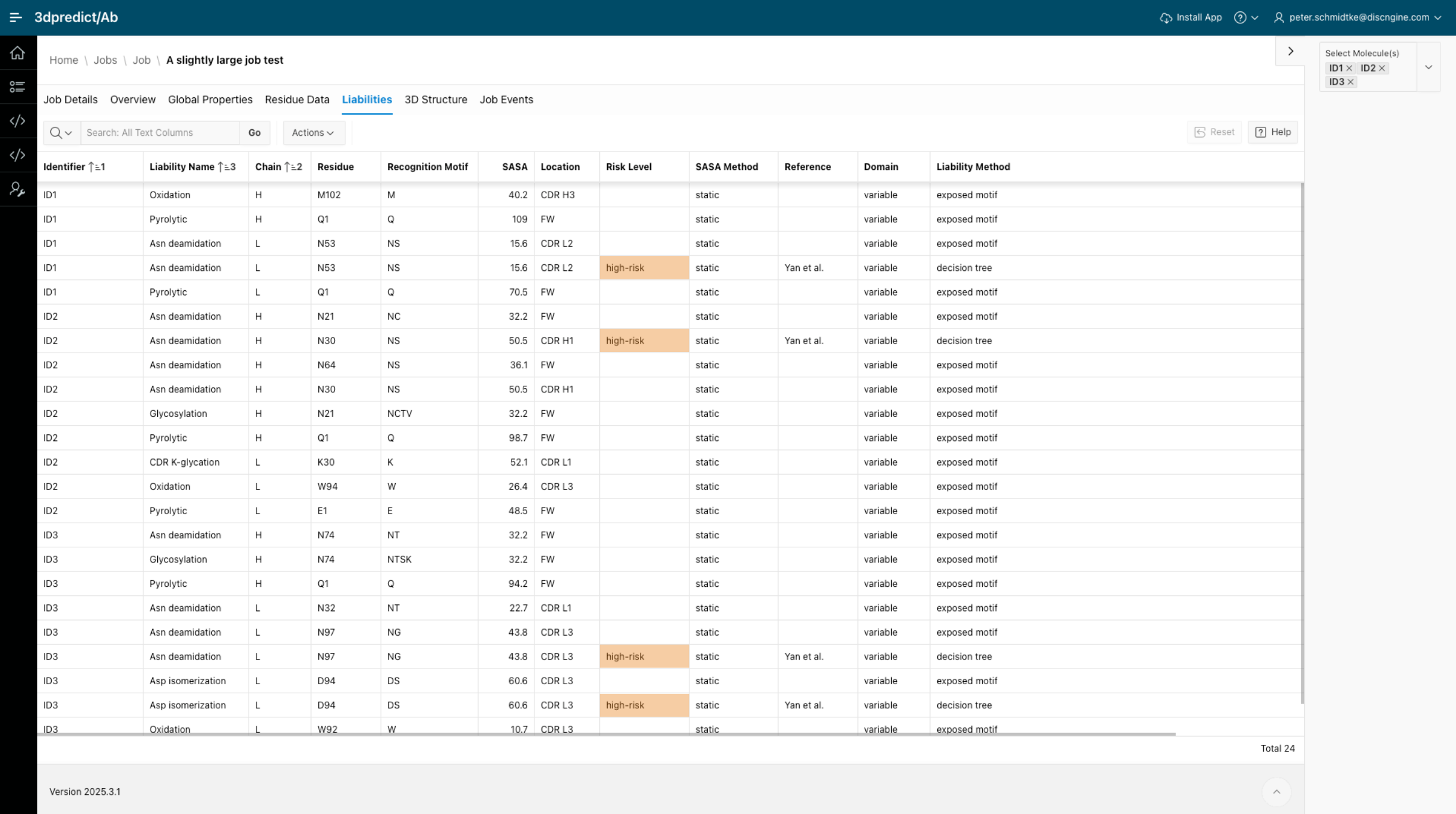
Accelerating early antibody discovery with ensemble-based developability assessment: 3dpredict/Ab
Discover how 3dpredict/Ab tackles current challenges in early antibody discovery by predicting hidden liability risks in candidates through an ensemble-based approach. By looking beyond traditional methods, it provides insights that help researchers make faster, more informed decisions on their candidates.

Highlights from Discngine Meetup Vol. 5: Pushing boundaries in peptide discovery with innovative science and technology
Discngine Meetup celebrated its fifth edition and brought together drug discovery experts to discuss recent progress and challenges in peptide discovery and design. Access event highlights and recordings.

“RNA as a Drug Target” Book Highlight: How targeting RNA emerges as the next frontier for medicinal chemistry
Our scientific expert Riccardo Martini shared an extensive overview of the famous book “RNA as a drug target”, incorporating his thoughts from the modelers' and the medicinal chemists’ perspectives.