Structure-based lead optimization of a PROTAC small-molecule in the BRD4-CRBN complex
As part of my learning experience, I experimented on how 3decision can support structure-based lead optimization projects. This time, the goal of my small experiment was to find a way to potentially improve the binding of the small molecule degrader dBET23 to the first bromodomain of the bromodomain-containing protein 4 – E3 ubiquitin ligase cereblon complex (BRD4 BD1-CRBN). Some of the features I used to generate and test design ideas, such as Subpocket Similarity search and Docking analysis with protein-ligand interaction coloring statistics, are quite unique to 3decision and probably new also for you, so make sure to stay with me throughout the whole project description.
In case you missed it, have a look at my previous article where I described how you can identify and characterize druggable binding sites on a protein surface with 3decision Pocket Explorer.
Introduction
The BRD4 belongs to the bromodomain and extra-terminal (BET) protein family together with BRD2, BRD3 and testis-specific BRDT. All members have two bromodomains (BD1 and BD2) and an extra-terminal domain (ET). The BRD4 bromodomains bind to acetylated lysins on histones and are a part of larger protein-protein complexes which activation results in transcriptional regulation. Dysfunction of BRD4 is associated with the development of solid tumors and hematological malignancies (reference here).
In fact, the inhibition of BRD4 by means of small drug-like molecules has been an effective anticancer therapy. A considerable number of BRD4 inhibitors have been developed, of which some are currently undergoing clinical trials. Common issues observed in trials, such as poor clinical response and off-target effects, imply the need for improvement. One of the alternative approaches is the use of proteolysis targeting chimeras (PROTACs) technology to degrade BRD4 and BET proteins in general. Many studies have shown that BRD4 degraders are more efficient anticancer agents than classical protein inhibitors (reference here).

Image 1: Chemical structure of dBET23 compound degrader, reference here
In this use case, I investigated a crystal structure where the PROTAC dBET23 is bound to BDR4 BD1 and the E3 ubiquitin ligase component cereblon CRBN, PDB ID: 6BN7 (Image 1). The PROTAC molecule is composed of a known pan-BET-inhibitor JQ1 as the target-moiety, an alkyl linker and a thalidomide E3-moiety. The goal of this theoretical experiment was to modify dBET23’s target-moiety with the aim to improve its binding affinity for BRD4’s first bromodomain.
Subpocket Similarity Search
By visual analysis of the pocket surface with a mesh representation, I noticed that the methyl group on the 1,2,4 - triazole of JQ1 points directly towards an empty binding site region (Image 2). Such an area appears to be able to accommodate further functional groups that might increase the binding affinity of the JQ1 inhibitor.
But how and with which functional group could we grow the PROTAC molecule?

Image 2: Visual inspection of an empty binding site subregion.
To get options on how to modify the dBET23 in the defined region, I used 3decision to perform a Subpocket Similarity search. It is an exclusive analytical tool that allows me to search targets that have similar subregions of the binding site. This way, I can have more insights into the chemical matter that binds to similar environments in other structures in the database and get ideas of potential modifications for dBET23.

Image 3: Selected residues for Subpocket Similarity Search.
All I needed at this point was to define the subregion of interest by selecting a set of residues around my ligand atom and launch the search (Image 3).
After a couple of minutes, 3decision retrieved a list of 531 structures and 491 co-crystallized ligands containing a similar subregion of the binding site, sorted by similarity score. The majority of the hits obtained were structures of BRD4 (target protein) with co-crystalized ligands.
I then studied the hit ligand library by superposing structures to my query. It was very interesting to see that a couple of BRD4 structures (PDB IDs: 6WGX, 4O7F, 5I88), as well as one BRD2 structure (PDB ID: 6MO7), had different functional groups occupying the selected binding site space where I wanted to grow my compound (Images 4 and 5).
These substituents coming from the same and/or even from the different targets might indeed serve as a starting point for modifications.
In further text, the selected substituents will be termed as follows:
Functional group A - p-fluorophenyl group
Functional group B - but-2-en-1-yl group
Functional group C - p-trifluoromethyl-phenyl group
Functional group D - N-methylacetamide

Image 4: Selected ligands from Subpocket Similarity search belonging to BRD4 which functional groups (depicted in green) occupy investigated binding site and could serve as a starting point for PROTAC modification.

Image 5: Ligand belonging to BRD2 -functional group depicted in green considered for modification.
Moreover, two of these compounds (PDB IDs: 6WGX, 4O7F) had been designed and tested to have preferred binding towards BRD4 BD1 with respect to BD2 (reference here). Functional groups A and B were added to replace the crystallographic water in the subregion I queried. Targeting crystallographic water has been proposed by Wellaway et al. to help increase BD1 affinity and selectivity. Therefore, I will modify the warhead of dBET23 with a functional group capable of displacing the crystallographic water and potentially increase the affinity towards the BRD4 D1 target moiety.
Ligand Docking and Interaction Scoring Feature
Once I got ideas on how I could make a modification on the JQ1 (BRD4 warhead), I designed a couple of derivatives and redocked them into the binding site. To facilitate the docking result analysis, 3decision has an interesting and unique visual representation of protein-ligand contact statistics. Each atom on the ligand in contact with the target is color-coded by the frequency of its occurrence in the same ligand-protein environment in other experimentally resolved structures. The green color represents a very common protein-ligand contact in other complexes in the 3decision database, or in other words – a very possible or relevant position. The colors from yellow to orange to red indicate that the position is less frequent. This feature can help us to rank docking poses and thereby prioritize designed compounds for synthesis.


Image 6 - Representation of the functional groups found through the Subpocket Similarity Search and added to the PROTAC warhead JQ1
Among the compound ideas I have sketched, the first was an addition of Functional group A on the JQ1. Such compound alteration had the atom coloring dominantly yellow in the studied binding site area (Image 7). Also, the modification with the functional group B showed a favorable position with the dominant green color (Image 8). Protein-ligand interaction fit adding functional group C seems worse than the others, probably because this group is too bulky. Therefore, the results showed “exotic” coloring on the trifluoromethyl group (Image 9). On the other hand, the functional group D coming from a different protein (BRD2) than the primary target (BRD4) allows the compound to keep the same binding mode (Image 10).

Image 7 - Docking results for compound A
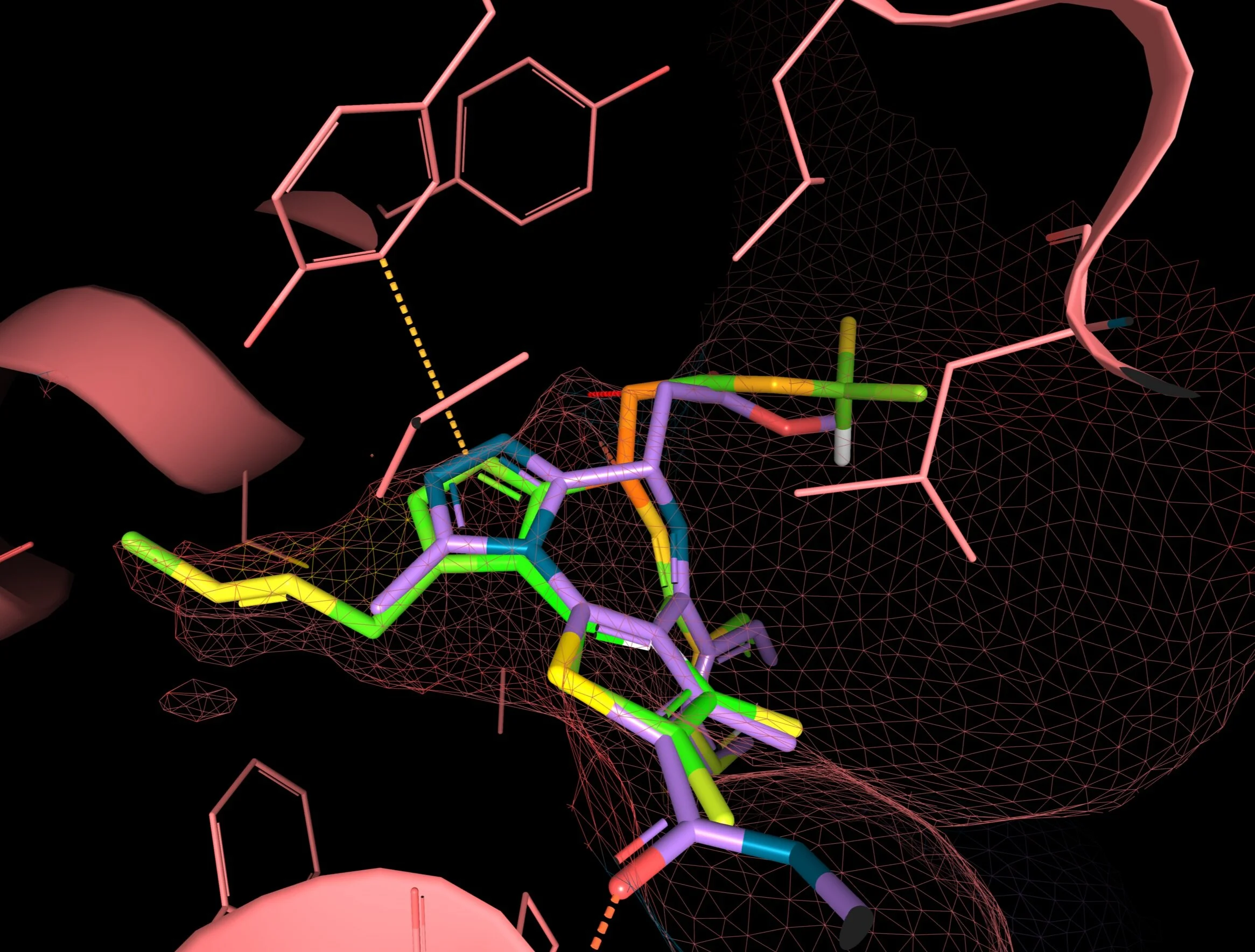
Image 8 - Docking results for compound B

Image 9 - Docking results for compound C

Image 10 - Docking results for compound D
Summary
In this theoretical exercise, I generated a hypothesis for the improved affinity of the dBET23 PROTAC compound towards the BRD4 BD1-CRBN complex. Based on structural analysis, I filled in an investigated pocket region on the PROTAC target moiety with modifications inspired by Subpocket Similarity Search results. This state-of-the-art search algorithm enabled me to find similar subregions of the binding site not only in other structures of my target but also in other proteins and to get an idea of which other chemical matter binds to similar environments. Moreover, the unique protein-ligand interaction coloring by statistics helped me to quickly assess whether the docking poses of new compounds were realistic compared to their occurrence in other protein-ligand complexes in the database and thus prioritize compounds for synthesis.
To experience more about 3decision and its functionalities, visit 3decision website here and follow us on social media for the latest news and updates.